
Abstract
Theefficient two-step synthesis of 2-chloro-2′-deoxyadenosine (cladribine) via the anion glycosylation of purine potassium salt with the glycosyl chloride in binary solvent mixtures is described. A new method for preparation of diprotected 2-chloro-6-fluoropurine 2′-deoxy-β- D-riboside was developed by treatment of the 2,6-dichloropurine precursor with diethylaminosulfur trifluoride (DAST). Novel N6-alkylated cladribine analogue was synthesized by amination of acylated 2,6-dihalogenopurine nucleosides. It was found that a mild hydrolysis reaction of acylated 2-chloro-6-fluoropurine 2′-deoxy-β-D-riboside gave rise to new purine hydroxylated nucleoside.
Author Contributions
Academic Editor: Jie Xu, Wuhan Textile University.
Checked for plagiarism: Yes
Review by: Single-blind
Copyright © 2021 Grigorii Sivets, et al.
 This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Competing interests
The authors have declared that no competing interests exist.
Citation:
Introduction
Among a series of known antineoplastic agents belonging to the purine nucleosides, cladribine have found application as the clinical drug for the treatment of hematologic malignances. The cladribine (2-chloro-2′-deoxyadenosine), deaminase-resistant analogue of 2′-deoxyadenosine is used for monotherapy of patients with hairy cell leukemia 1 and treatment of other lymphoid malignances. Its mechanism of significant cytotoxicity and metabolism was widely studied and it has been established that the active metabolite, 5′-triphosphate, inhibits DNA synthesis and ribonucleotide reductase activity 2, 3. Recently, cladribine has also been approved as the oral drug with a promising efficacy and safety profile 4 for the treatment of relapsing multiple sclerosisin adults 5.
The synthesis of cladribine has widely been investigated in the framework of several approaches a) glycosylation reactions of purine derivatives with sugars 6, 7, 8, 9; b) C2′-deoxygenation of selectively protected purine nucleoside derivative 10; c) enzymatic transglycosylation or glycosylation reactions 11, 12, 13. It should be noted, of the known synthetic approaches, the most studied method is derived from glycosylation reactions of purine nucleobase derivative with an activated carbohydrate. The sodium salts of halogenated purines give N9 and N7 glycosyl isomers under the glycosylation and the stereoselectivity varies with heterocyclic bases and the reaction conditions 12, 13.This challenge was solved via highly stereoselective glycosylation of 6-substituted imidazol-1-yl-2-chloropurines with protected 1-chloro-2-deoxyribose in a mixture of solvents, using Robins’ purine salt method, and cladribine was prepared in three steps from the N6-modified purine 14. A practical and efficient process for the manufacture of cladribine was developed via the Vorbrüggen glycosylation of silylated base with 1-acetoxy 2-deoxyribofuranose derivative 11. N6-alkylated purine 2′-deoxyribonucleoside analogues display in vitro anticancer and antiviral activities 15, 16. Because of our interest in extending preparation of biologically active nucleosides from sugars, herein we report study of efficient two-step synthetic route to 2-chloro-2′-deoxyadenosine via stereoselective anion glycosylation of purine base with the carbohydrate precursor using readily available reagents and development of simple synthetic approaches to its novel purine modified analogues.
Results and Discussion
Synthesis of cladribine 3 was explored via selective glycosylation of the potassium salt of 2,6-dichloropurine with available 1-α-chlorosugar 1 17 using various reaction conditions on the key step to increase its regioselectivity (Scheme 1, Table 1). Regioselective coupling of the potassium salt of 2,6-dichloropurine, generated in the presence of potassium tert-butoxide in 1,2-dimethoxyethane, with 1-chloro-2-deoxy-3,5-di-O-p-toluoyl-α-D-erythro-pentofuranosyl chloride (1) at ambient temperature in a mixture of anhydrous acetonitrile and tetrahydrofuran resulted in formation of 3′,5′-di-O-toluoyl-2,6-dichloropurine-2′-deoxy-β-D-riboside (2) as predominant product. 1H NMR data of the crude reaction mixture indicate that glycosylation proceeds with full conversion of the starting 1-α-chlorosugar to a mixture of three nucleosides, giving acylated N9-β-2′-deoxy-β-D-riboside along with the undesired N9-α- and N7-β-isomeric nucleosides as by-products. A ratio of acylated N9-β/ N7-β-regioisomers made up 6.2:1 (Table 1, entry 1). Protected N9-2′-deoxy-β-D-nucleoside 2 was prepared in 70% yield after column chromatography on silica gel. The anion glycosylation reaction of 2,6-dichloropurine salt in anhydrous tetrahydrofuran in the presence of 18 crown 6 resulted in the intermediate N9-β-nucleoside 2 in 61% yield after chromatographic separation, N9-β/ N7-β-regioisomeric nucleosides being formed in a ratio of 5.2:1 (entry 2). The glycosylation reaction in THF, the solvent with a lower polarity, and in the presence of crown ether as additive for improving solubility of the purine salt gave rise to decrease of the reaction time, yield of the protected N9-β-nucleoside and stereoselectivity compared to the reaction in a binary mixture of solvents (Scheme 1, conditions a1 and a2).Next,the glycosylation reaction of the potassium salt of 2,6-dichloropurine in a binary solvent mixture (acetonitrile and 3,4 dihydro-2H-pyran as component with lower polarity) gave the best regioselectivity (ratio of protected N9-β/ N7-β-/N9-α-nucleosides ‒ 10:1:1)(entry 3). The protected N9-β-nucleoside 2 was isolated in 67-70%yield after column chromatography on silica gel using mixtures of petroleum ether and ethylacetate. The above results on the anion glycosylation of the 2,6-dichloropurine salt with the 1-α-chlorosugar under tested conditions provide evidence for regio- and stereoselectivity of the reaction depends on rational choice of solvents with different solvation of the purine potassium salt and a minimal anomerization of the starting sugar. Fast anion glycosylation of the purine potassium salt with the 1-α-chlorosugar in binary solvent mixtures improves the stereoselectivity of the heterogeneous reaction to give higher isolated yield of acylated N9-β-nucleoside than the sodium salt glycosylation in acetonitrile 7. Protected 2′-deoxy-β-D-ribonucleoside of 2,6-dichloropurine 2 was then converted to cladribine 3 in NH3/MeOH/THF. Selectiveammonolysis of 2 occurring with the deprotection gave 2-chloro-2'-deoxyadenosine (3) in 82% yield after column chromatography. The improved approach to cladribine via coupling of the potassium salt of 2,6-dichloropurine with the 1-α-chlorosugar in a mixture of solvents was accomplished using commercially available reagents. It should be noted that overall yields (50-56%) of the target nucleoside are comparable to those for efficient chemical approaches (42-63%) developed from 2-deoxy-D-ribofuranose derivatives 6, 7, 9, 14 and 2-chloroadenosine 10, or the enzymatic method (59%) described from thymidine 13. However, inherent drawbacks of the studied method are the formation of N-9- and N-7-isomers on the glycosylation step, unlike the enzymatic methods 11, 12, 13, and the use of chromatography on two steps in comparison with the cost-efficient and practical four-step procedure via the Vorbrüggen glycosylation of silylated 2-chloroadenine (overall 42%) 6. When this method is compared with the known synthetic routes derived from 2,6-dichloropurine and 2-deoxy-D-ribofuranose derivatives it is apparent that cladribine synthesis via the glycosylation of the potassium purine salt in a binary mixture of solvents and ammonolysis under mild reaction conditions (Scheme 1) produced higher overall yield (56%) than the sodium salt method (42%) 7. Besides, the studied two-step approach is more accessible than the five-step method reported earlier via the stereoselective glycosylation of 2,6-dichloropurine with peracylated 2-deoxy-D-ribofuranose in the presence of gold-containing catalyst 9 and less efficient than Robins’ purine salt method 14.
Scheme 1.Syntheses of cladribine 3 from 1-α-chlorosugar 1
| Entry | Solvent | Conditions Ratio of heterobase: chlorosugar | Time (min) | Anomeric ratios (N-9-β:N7-β)a | Yield (%)b |
| 1 | MeCN/THF (1:1.2) | 1.1:1 | 150 min | 6.2:1 | 70 |
| 2 | THFc | 1.1:1 | 120 min | 5.2:1 | 61 |
| 3 | MeCN/3,4-DHP(1:1.1) | 1.1:1 | 150 min | 10.0:1 | 67-70 |
Synthesis of new N6-alkylated derivative of cladribine containing a branched alkyl substituent was investigated from 3′,5′-di-O-p-toluoyl-2′-deoxy-β-D-riboside of 2,6-dichloropurine 2. Several synthetic approaches to biologically active N6-substituted purine nucleosides were described earlier 18 and the most employed methods are based upon introduction of a leaving group (halogen, aryl sulfonate, or O-benzotriazolyl) at the C-6 position of nucleoside derivative followed by a selective nucleophilic SNAr displacement with the corresponding alkyl amines 15. Fluorination of O-protected 2,6-dichloropurine 2′-deoxy-β-D-ribonucleoside 2 was performed with an excess of DAST in a mixture of CH2Cl2/pyridine under mild heating (Scheme 2).
Scheme 2.Synthesis of N6-alkylated cladribine derivative 6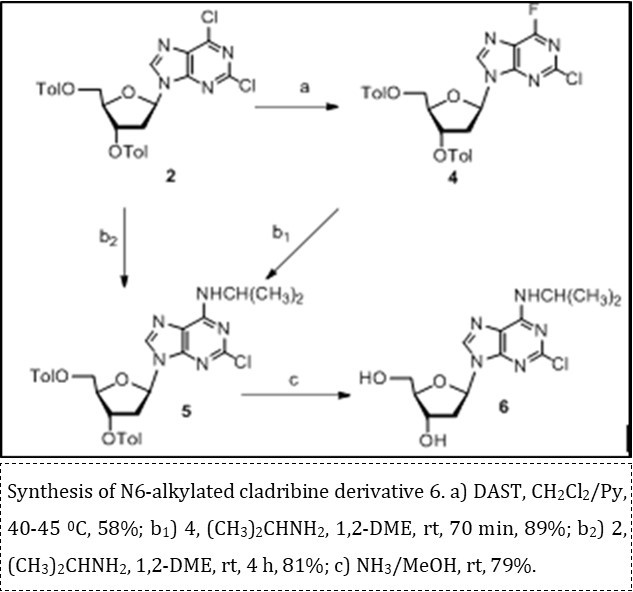
We have found for the first time that such treatment of protected 2,6-dichloropurine 2´-deoxyriboside by the nucleophilic fluorinating reagent gave rise to 2-chloro-6-fluoropurine derivative 4 in 58% yield after chromatography on silica gel. Syntheses of 6-fluorinated purine nucleosides and heterocyclic bases have earlier been reported in several works 19, 20, 21. Selective fluorination reaction of 2 likely to proceed by two pathways A and B via formation of activated intermediates 7, 8, and 9 followed by nucleophilic SNAr 6-chlorine displacement with DAST in adducts 7 and 9 (Scheme 3). Based on a previous study of synthesis of 6-fluorinated purines and the known method 21 developed by Deng et al., we can conclude that pathway A via formation of intermediate salt 7 is a predominant.
Scheme 3.Proposed mechanism for fluorination reaction of 2,6-dichloropurine nucleoside 2 with DAST
Further, we investigated comparable amination of protected 2-chloro-6-fluoropurine (4) and 2,6-dichloropurine (2) 2′-deoxy-β-D-ribosides with isopropyl amine in similar reaction conditions. Reactions of 2,6-dihalogenated purine derivatives 2 and 4 using fourfold excess of diisopropylamine in 1,2-dimethoxyethane at room temperature resulted in N6-alkylated purine analogue 5 in 81% and 89% yield, respectively, after chromatography on silica gel (Scheme 2).1H NMR spectral data of N6-isopropyl purine derivative 5 synthesized from 2,6-dichloropurine and 2-chloro-6-fluoropurine derivatives were identical, that unequivocally confirms the assigned structure of new 2-chloro-6-fluoropurine nucleoside precursor 4. Noteworthy, a selective amination of 4 proceeded in higher yield and shorter reaction time than the same transformation of 2,6-dichloropurine derivative 2. Thus, protected 2-chloro-6-fluoropurine 2′-deoxy-β-D-riboside can be utilized as valuable intermediate for preparation of different N6 or C6-substituted purine 2′-deoxyribonucleosides of biological interest via SNAr selective mild displacement of fluorine atom by various nucleophilic agents. Deprotection of O-acylated N6-isopropyl purine nucleoside 5 with ammonia in methanol afforded novel cladribine analogue 6 in 79% yield after column chromatography on silica gel. Then, hydrolysis reaction of 2-chloro-6-fluoropurine 2′-deoxy-β-D-ribofuranoside 4 was studied under mild basic treatment with sodium hydrocarbonate in acetonitrile (Scheme 4). New purine modified nucleoside 14 was prepared in 65% yield after chromatography on silica gel. A possible mechanism for formation of hydroxylated purine nucleoside 14 via SNAr selective displacement of fluorine atom in 4 on the first step and subsequent transformations of intermediate 2-chloro-3′,5′-di-O-p-toluoyl-2′-deoxyinosine (10) in the heterobase under mild basic conditions is outlined in Scheme 4. Structures of synthesized novel purine modified nucleosides were confirmed by 1H, 13C, 19F NMR and mass-spectroscopy. Signal of F-6 atom in 2-chloro-6-fluoropurine nucleoside 4 displays as a singlet at - 65.59 ppm in 19F NMR spectrum. Chemical shift of fluorine atom in 2-chloro-6-fluoropurine derivative 4 is in good accordance with 19F NMR spectral data of tri-O-acetylated 2-chloro-6-fluoropurine β-ribonucleoside described earlier 19. The presence of hydroxy groups at 3.9-4.5 ppm and absence of signal for NH proton (8.1-10.0 ppm) in 1H NMR spectrum and peak of M+Na+ in mass-spectrum support the assigned structure of hydroxylated purine nucleoside 14.
Scheme 4.Synthesis of purine modified 2′-deoxynucleoside 14 from 2-chloro-6-fluoropurine derivative 4
Conclusion
In summary, the improved cladribine synthesis was achieved using the 2,6-dichloropurine potassium salt in the coupling reaction with available 1-α-chlorosugar and subsequent mild ammonolysis of the intermediate nucleoside. Solvent effects on stereoselectivity and regioselectivity of the glycosylation procedure were established. A new method to synthesize diprotected 2-chloro-6-fluoropurine 2′-deoxy-β-D-ribofuranoside was developed for the first time from the 2,6-dichloropurine nucloside using nucleophilic fluorinating agent and a plausible mechanism for formation of 2-chloro-6-fluoropurine derivative was proposed by selective nucleophilic SNAr 6-chlorine displacement in the starting nucleoside with DAST. The efficient synthesis of N6-alkylated cladribine analogue was accomplished via a selective amination of 2-chloro-6-fluoropurine nucleoside derivative. New purine modified nucleoside derivative was prepared by hydrolysis reaction of acylated 2-chloro-6-fluoropurine 2′-deoxy-β-D-ribofuranoside and a possible mechanism for its formation was considered.
Materials and Methods
Column chromatography was performed on silica gel 60 H (70-230 mesh; Merck, Darmstadt, Germany), and thin-layer chromatography (TLC) on Merck silica gel aluminum 60 F254 precoated plates. The anhydrous solvents were distilled over CaH2, P2O5 or magnesium prior to the use. All commercially available reagents were used without further purification. 1H, 13C, and 19F NMR spectra were recorded in CDCl3, CD3OD and DMSO-d6 with a Bruker Avance-500-DRX spectrometer at 500.13, 126.76 and 470.59 MHz, respectively. 1H and13C NMR chemical shifts (δ, ppm) are relative to internal chloroform peak (7.26 ppm for 1H and 77.0 for 13C NMR). Chemical shifts are also reported downfield from internal SiMe4 (1H) or external CFCl3 (19F). Splitting patterns were reported as following: s: singlet, d: doublet, t: triplet, m: multiplet. J values are reported in Hz. Melting points were determined on a Boetius apparatus and were uncorrected. High resolution mass spectra (HRMS) were recorded on an Agilent Q-TOF 6550 Instrument (USA) using ESI (electrospray ionization).
Experimental Procedures
Synthesis of Cladribine 3, 5 and -2-
Synthesis of 2,6-dichloro-9-(3′,5′-di-O-p-toluoyl-2′-deoxy-β-D-ribofuranosyl)-9H-purine (2)
a1) Potassium t-butoxide (0.022 g, 0.18 mmol) was added to 2,6-dichloropurine (0.036 g, 0.19 mmol) in anhydrous 1,2-dimethoxyethane (3 ml) at 0 0C and then the resulting solution was stirred for 7 min under cooling and then for 15 min at room temperature and evaporated to dryness. Anhydrous acetonitrile (2.5 ml) and tetrahydrofuran (2.9 ml) were added to the residue and the suspension was stirred under argon at room temperature for 15 min, then the crystalline chloride 1 (0.07 g, 0.18 mmol) was gradually added during 10 min. The reaction mixture was stirred under argon at room temperature for 240 min. Insoluble materials were removed by filtration and the solids were washed with MeCN (5 mL). The solvent was removed under reduced pressure and the residue was chromatographed on silica gel, eluting with toluene/acetone 9:1 to afford β-nucleoside 2 as a white solid (0.068 g, 70%). Mp. 159-162 0С (EtOH).1H NMR (CDCl3, 500 MHz): δ 8.29 (s, 1Н, Н-8), 7.97, 7.82, 7.28, 7.20 (4d, 8Н, 2xTol), 6.55 (t, 1Н, J1′,2′= J1′,2′′ = 6.4 Hz, Н-1′), 5.79 (br.t, 1Н, Н-3′), 4.80 (dd, 1Н, JH-5′,H-4′= 2.7 Hz, JH5′,H5′′ = 10.5 Hz, Н-5′), 4.64-4.69 (m, 2Н, Н-4′ и Н-5′′), 2.94-2.97 (m, 2Н, Н-2′ and H-2′′), 2.45 (s, 3Н, CH3C6H4CO) and 2.41 (s, 3Н, CH3C6H4CO). 13C NMR (CDCl3, 126.76 MHz): δ 166.0 and 165.9 (СОC6H4CH3), 153.0, 152.2, 151.9, 144.6, 144.4, 143.7, 131.2, 129.8, 129.5, 129.4, 129.3, 126.3, 126.1 (С-6, С-2, С-4, С-5, С-8, 2xCH3C6H4CO), 85.3 (С-4′), 83.6 (С-1′), 74.9 (С-3′), 63.7 (С-5′), 38.5 (C-2′), 21.7 and 21.6 (2xCH3C6H4CO). HRMS (ESI) calcd for C26H22Cl2N4O5 (M + Na)+: 563,0865, found 563,0862.
a3) Potassium t-butoxide (0.029 g, 0.24 mmol) was added to 2,6-dichloropurine (0.052 g, 0.28 mmol) in anhydrous 1,2-dimethoxyethane (4 ml) at 0 0C and then the resulting solution was stirred for 7 min under cooling and then for 15 min at room temperature and evaporated to dryness. Anhydrous acetonitrile (4 ml) and freshly distilled 3,4-dihydropyrane (4.4 ml) were added to the residue and the suspension was stirred under argon at room temperature for 15 min, then the crystalline chloride 1 (0.1 g, 0.26 mmol) was gradually added during 10 min. The reaction mixture was stirred under argon at room temperature for 210 min. Insoluble materials were removed by filtration and the solids were washed with MeCN (20 mL). The solvent was removed under reduced pressure and the residue was chromatographed on silica gel, eluting with EtOAc/petroleum ether 3:1 and 2:1 to afford β-nucleoside 2 a white solid (0.094 g, 67%).
Synthesis of 2-chloro-2′-deoxyadenosine (3)
To a solution of β-nucleoside 2 (0.3g, 0.55 mmol)in anhydrous THF (14 mL) was added in 50 ml methanol saturated at 0 0C with ammonia, the reaction mixture was stirring for 24 h at room temperature, then for 18 h under 35-40 0C and evaporated. The residue was chromatographed on silica gel using for elution CHCl3,then CHCl3:MeOH-15:1 and 5:1 to afford nucleoside 3 as a white solid(0.13 g, 82%).Mp. >300 0С (methanol).1H NMR (DMSO-d6, 500 MHz): δ 8.36 (s, 1Н, Н-8), 7.82 (br. s , 2Н, NH2), 6.27 (t, 1Н, J1′,2′= J1′,2′′ = 6.4 Hz, Н-1′), 5.40 (d, 1Н, J3′,3-ОН = 4.2, 2′-ОН), 5.04 (t, 1Н, J5′,5-ОН = 5.4, 5′-OH), 4.10 (m, 1Н, Н-3′), 3.87 (m, 1Н, Н-4′). 3.53-3.62 (m, 2Н, Н-5′ and Н-5′′), 2.61 (ddd, 1Н, Н-2′), 2.32 (ddd, 1Н, Н-2′′). 13C NMR (DMSO-d6, 126.76 MHz): δ 156.8, 153.3, 150.1, 139.9, 118.2, 87.9, 83.6, 70.7, 61.7, 39.8. HRMS (ESI) calcd for C10H12ClN5O3: 286.0707 (M+H), found 286.0701.
Synthesis of Purine Modified Nucleoside Analogues 4, 5, 6 and 14
Synthesis of 2-chloro-6-fluoro-9-(3′,5′-di-O-p-toluoyl-2′-deoxy-β-D-ribofuranosyl)-9H-purine (4).
To a solution of 3′,5′-di-O-toluoyl-2,6-dichloropurine-2′-deoxyriboside (2) (0.06 g, 0.11 mmol) in anhydrous CH2Cl2 (3.7 ml) and pyridine (0.022 ml) was added dropwise 0.095 ml (0.72 mmоl) DAST at room temperature. The reaction mixture was stirred for 30 min at rt and 23 h at 40-45 0C, then evaporated. The residue was chromatographed on silica gel using for elution mixtures 7:1, 5:1 and 3:1 hexane-EtOAc. Nucleoside 4 (0.034 g, 58%) was prepared as a syrup. Mp. 142-143 0С (ethylacetate/hexane).1H NMR (CDCl3, 500 MHz): δ 8.32 (s, 1H, H-8), 8.02 (d, 2H, Tol), 7.88 (d, 2H, Tol), 7.33 (d, 2H, Tol), 7.25 (d, 2H, Tol), 6.61 (t, 1H, J= 5.9 Hz, J = 6.9 Hz, H-1′), 5.82-5.86 (m, 1H, H-3′), 4.83 (dd, 1H, JH-5′,H-4′ = 6.8 Hz, JH5′,H5′′ = 11.0 Hz, H-5′), 4.67-4.75 (m, 2H, H-4′ and H-5′′), 2.98-3.05 (m, 2H,H-2′ and H-2′′), 2.49 (s, 3H, Tol), 2.45 (s, 3H, Tol). 13C NMR (CDCl3,126.76 MHz): δ 166.1 and 166.0 (C=O, Tol), 159.3 (d, J = 265.0 Hz), 155.9 (d, J = 11.2 Hz), 152.7 (d, J = 16.6 Hz), 144.8, 144.5, 143.5 (d, JC-8,F-6 ~ 2.0 Hz, C-8), 129.9, 129.6,129.4, 126.4, 126.2, 119.7, 85.4 (C-4′), 83.7 (C-1′), 74.9 (C-3′), 63.8 (C-5′), 38.6 (C-2′), 21.8 (CH3C6H5CO-), 21.7 (CH3C6H5CO-). 19F NMR (CDCl3, 470.59 MHz): δ -65.59 (s, F-6). HRMS (ESI) calcd for C26H22ClFN4O5M+Na+: 547.1160, found 547.1157.
Synthesis of 2-chloro-6-isopropylamino-9-(3′,5′-di-O-p-toluoyl-2′-deoxy-β-D-ribofuranosyl)-9H-purine (5)
To a solution of 3′,5′-di-O-toluoyl-2-chloro-6-fluoropurine-2′-deoxyriboside (4) (0.018 g, 0.0034 mmol) in anhydrous 1,2-DME(2.0 ml) was added 0.013 ml (0.17 mmоl) isopropyl amine at room temperature. The reaction mixture was stirred for 70 min and then evaporated. The residue was chromatographed on a silica gel, using a mixture of 4:1, 2:1, 1:1 and hexane-EtOAc to affordnucleoside 5(0.017 g, 89%)as a syrup. Mp.72-74 0С (MeOH).1H NMR (CDCl3, 500 MHz): δ 8.02 (d, 2H, Tol), 7.97 (s, 1H, H-8), 7.93 (d, 2H, Tol), 7.33 (d, 2H, Tol), 7.27 (d, 2H, Tol), 6.56 (t, 1H, J1′,2′= J1′,2′′ = 6.4 Hz, H-1′), 6.02 (br. s, 1H, NH), 5.75-5.83 (m, 1H, H-3′), 4.78 (dd, 1H, JH-5′,H-4′ = 3.2 Hz, JH5′,H5′′ = 11.0 Hz, H-5′), 4.69 (dd, 1H, JH-5′′,H-4′ = 3.3 Hz, H-5′′), 4.63-4.70 (m, 1H, H-4′), 4.52 [br. s, 1H, HNCH(CH3)2], 2.90-2.93 (m, 2H,H-2′ and H-2′′), 2.49 (s, 3H, Tol), 2.45 (s, 3H, Tol), 1.35 [d, 3H, J = 0.9 Hz, CH(CH3)2], 1.34 [d, 3H, J = 0.9 Hz, CH(CH3)2]. 13C NMR (CDCl3,126.76 MHz): δ 166.2 and 166.0 (C=O, Tol), 154.5, 144.6, 144.3, 137.4, 129.9, 129.6,129.4, 129.3, 126.7, 126.4 (C-6, C-2, C-4, C-8, C-5, CH3C6H5CO-), 84.6 (C-4′), 83.2 (C-1′), 75.1 (C-3′), 64.1 (C-5′), 42.8 [CH(CH3)2], 38.7 (C-2′), 22.8 [CH(CH3)2], 21.8 (CH3C6H5CO), 21.7 (CH3C6H5CO).HRMS (ESI): m/z calcd for C29H30N5O5Cl (M+Na)+: 586.1883, found 586.1878.
Synthesis of 2-chloro-6-isopropylamino-9-(2′-deoxy-β-D-ribofuranosyl)-9H-purine (6)
A solution of nucleoside 5 (0.034 g, 0.06 mmol)in MeOH (4 mL) saturated at 0 0C with ammonia was kept for 18 h at room temperature and then evaporated. The residue was chromatographed on silica gel, eluting with CHCl3,then CHCl3:MeOH-20:1, 15:1 and 8:1 to afford nucleoside 6 (15.6 mg, 79%). Mp.124-126 0С (MeOH).1H NMR (CD3OD, 500 MHz): δ 8.25 (s, 1H, H-8), 6.39 (dd, 1H, J1′,2′ = J1′, 2′′ = 6.2 Hz, H-1′), 4.58-4.61 (m, 1H, H-3′), 4.43 [br. s, 1H, HNCH(CH3)2], 4.07 (q, 1H, H-4′), 3.87 (dd, 1H, J5′,4′ = 3.2 Hz, J5′,5′′ = 12.2 Hz, H-5′), 3.76 (dd, 1H, J5′′,4′ = 3.6 Hz, H-5′′), 2.87 (ddd, 1H, J = 5.5 Hz, J = 6.2 Hz, J = 12.3 Hz, H-2′), 2.42 (ddd, 1H, J = 2.7 Hz, J = 6.2 Hz, H-2′′), 1.33 [br.s, 1H, CH(CH3)2], 1.31 [br.s, 1H, CH(CH3)2]. 13C NMR (CD3OD, 126.76 MHz): δ 155.9, 155.6, 150.6, 120.1, 89.8, 87.0, 72.9, 74.8, 63.6, 43.8, 41.5, 22.7. HRMS (ESI): m/z calcd for C13H18N5O3Cl M+Na+: 350.0996, found 350.0983.
Synthesis of 2-chloro-4,5-dihydroxy-3′,5′-di-O-p-toluoyl-2′-deoxyinosine (14).
To a solution of 3′,5′-di-O-toluoyl-2,6-dichloropurine-2′-deoxyriboside (2) (0.06 g, 0.11 mmol) in anhydrous CH2Cl2 (3.7 ml) and pyridine (0.022 ml) was added dropwise 0.095 ml (0.72 mmоl) DAST at room temperature. The reaction mixture was stirred for 30 min and 23 h at 40-45 0C, then evaporated. To the residue was added 4.0 ml aq. NaHCO3 and acetonitrile (1.2 ml) and reaction mixture was stirred at rt for 24 h. then water phase was extracted with CHCl3(3x20ml), combined organic layer was washed with water and dried over sodium sulfate. The residue was chromatographed on silica gel, using for elution mixtures of 6:1, 5:1 and 3:1 petrolium ether-EtOAc to afford nucleoside 14 (0.023 g, 65%) as oil. 1H NMR (CDCl3, 500 MHz): δ 8.03 (s, 1H, H-8), 8.02 (d, 2H, Tol), 7.88 (d, 2H, Tol), 7.33 (d, 2H, Tol), 7.26 (d, 2H, Tol), 6.59 (t, 1H, J1′,2′= J1′,2′′ = 6.4 Hz, H-1′), 5.77-5.80 (m, 1H, H-3′), 4.75 (dd, 1H, JH-5′,H-4′ = 3.3 Hz, JH5′,H5′′ = 11.0 Hz, H-5′), 4.71 (dd, 1H, JH-5′′,H-4′ = 4.1 Hz, H-5′′), 5.65-5.67 (m, 1H, H-4′), 4.2 (br.s, 1.6H, OH), 3.9 (br.s, 1.4H, OH), 2.91-2.93 (m, 2H,H-2′ and H-2′′), 2.49 (s, 3H, Tol), 2.45 (s, 3H, Tol). 13C NMR (CDCl3, 126.76 MHz): δ 166.2 and 166.0 (C=O, Tol), 154.3, 153.8, 151.1, 144.5, 144.2, 136.2 129.9, 129.7,129.3, 126.7, 126.4, 118.2, 84.5 (C-4′), 83.1 (C-1′), 75.1 (C-3′), 64.2 (C-5′), 38.6 (C-2′), 21.8 (CH3C6H5CO-), 21.7 (CH3C6H5CO-). LS (ESI) calcd for C26H25ClN4O8M+Na+: 578.7, found 578.7.
Acknowledgments
This study was supported by grant from FOI «Chemical processes, reagents and technologies, bioregulators and bioorgchemistry», s/p «Chemical foundations of life activity processes» (Bioorgchemisrty 2.3.2.2).
References
- 1.Inwards D, Fishkin P, Laplant B, Drake M, Kurtin P. (2014) Phase I trial of rituximab, cladribine and temsirolimus (RCT) for initial therapy of mantle cell lymphoma. , Ann.Oncol 25(10), 2020-2024.
- 2.Shelton J, Lu X, Hollenbaugh J, Cho J, Amblard F. (2016) Metabolism biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs. , Chem. Rev 116(23), 14379-14455.
- 3.Robak T, Korycka A, Lech-Maranda E, Robak P. (2009) Current status of older and new purine nucleoside analogues in the treatment of lymphoproliferative diseases. , Molecules 14, 1183-1226.
- 4.Schreiber K, Sorensen P. (2011) Cladribine treatment of multiple sclerosis. , Medicine. Clin. Invest 1(2), 317-326.
- 5.Mulakayata N, Rao P, Iqbat J, Bandichhor R, Oruganti S. (2013) Synthesis of novel theraupetic agents for the treatment of multiple sclerosis. A brief overreview. , Eur. J. Med. Chem 80, 170-186.
- 6.Henschke J, Zhang X, Huang X, Mei L, Chu G. (2013) A stereoselective process for the manufature of a 2′-deoxy-β-D-ribonucleoside using the Vorbruggen glycosylation. , Org. Process Res.& Dev 17, 1419-1429.
- 7.Kazimierczuk Z, Cottam H, Revamkar G, Robins R. (1984) Synthesis 2-deoxyrtubercidin, 2′-deoxyadenosine and related 2′-deoxynucleosides via a novel direct sterospecific sodium salt glycosylation procedure. , J.Am.Chem.Soc 106(21), 6379-6382.
- 8.Hildebrand C, Wright G. (1992) Sodium salt glycosylation in the synthesis of purine 2-deoxyribonucleosides: studies of isomer distribution. , J.Org.Chem 57(6), 1808-1813.
- 9.Yang F, Zhu Y, Yu B. (2012) A dramatic concentration effect on the stereoselectiveity of N-glycosylation for the synthesis of 2′-deoxy-β-ribonucleosides. , Chem.Commun 48, 7097-7099.
- 10.Xia R, Chen L. (2015) Efficient synthesis of cladribine via the metal-free deoxygenation. , Nucleosides, Nucleotides Nucleic Acids 34, 729-735.
- 11.Mikhailopulo I, Zinchenko A, Kazimierczuk Z, Barai V, Bokut S. (1993) Synthesis of 2-chloro 2′-deoxyadenosine by microbiological trans glycosylation. , Nucleosides, Nucleotides12(3-4): 417-422.
- 12.Komatsu H, Araki T. (2005) Efficient chemo-enzymatic syntheses of pharmaceutically useful unnatural 2′-deoxynucleosides, Nucleosides, Nucleotides Nucleic Acids24(5-7):. 1127-1130.
- 13.Artemyeva J, Remeeeva E, Buravskaya T, Konstantinova I, Esipov R. (2020) Anion exchange resins in phosphate form as versatile carriers for the reactions catalyzed by nucleoside phosphorylases. , Beilstein J. Org. Chem 26, 2607-2622.
- 14.Znong M, Novak L, Robins M. (2006) Regiospesific and highly streoselective coupling of 6-(substituted-imidazol-1-yl)purines with 2-deoxy-3,5-di-O-(p-toluoyl)-D-erythro-pentofuranosyl chloride. Sodium-salt glycosylation in binary solvent mixtures: improved synthesis of cladribine. , J. Org. Chem 71(20), 7773-7779.
- 15.Satishkumar S, Vuram P, Relangi S, Gurram V, Zhou H. (2015) Cladribine analogues via O6-(benzotriazolyl) derivatives of guanine nucleosides. , Molecules 20, 181-202.
- 16.Salvatori D, Volpini R, Vincenzetti S, Vita A, Costanzi S. (2002) Adenine and deazaadenine nucleoside and deoxynucleoside analogues: inhibition of viral replication of sheep MVV (in vitro model for HIV) and bovine BHV-1. , Bioorg. Med. Chem 10, 2973-2980.
- 17.Rolland V, Kotera M, Lhomme J. (1997) Convenient preparation of 2-deoxy-3,5-di-O-p-toluoyl-α-D-erythro-pentofuranosyl chloride. , Synth. Commun 27(2), 3505-3511.
- 18.Drenichev M, Oslovsky V, Mikhailov S. (2016) . Cytokinin Nucleosides – Natural Compounds with a Unique Spectrum of Biological Activities, Current Topics in Med. Chem.16 2562-2576.
- 19.Robins M, Yznanski B. (1981) Nucleic acid related compounds. 34. Non-aqueous diazotization with tert-butyl nitrite. Introduction of fluorine, chlorine, and bromine at C-2 purine nucleosides. , Can J. Chem 59, 2608-2611.